
Schlaepfer Lab
- Research
- Lab Personnel
- Publications
- Contact Us
We are funded by two NIH R01 grants from the National Cancer Institute: 1) to study molecular mechanisms of ovarian cancer drug resistance and 2) the role of FAK signaling within ovarian tumor cells leading to an immunosuppressive tumor microenvironment.
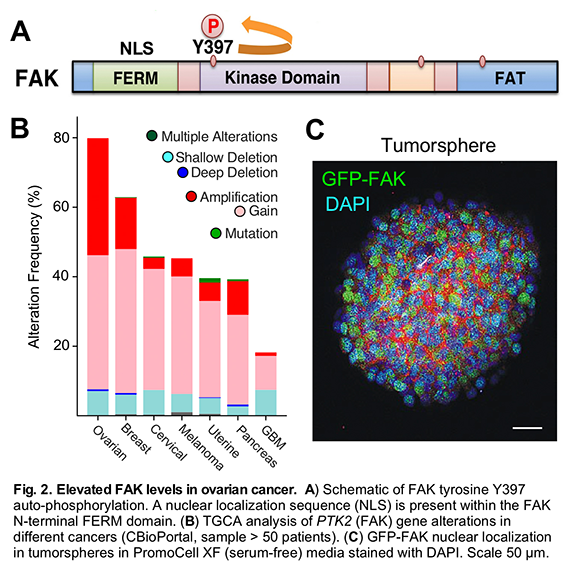
 PROJECT 1 – We have inactivated the FAK gene by CPRISPR DNA editing within murine and human ovarian tumor cells and have re-expressed wildtype or various function-altering mutations of FAK. The goal of these studies is to understand the role of FAK activity (it is an enzyme that will modify other proteins) as a function of its localization in cells. FAK is hypothesized to function at the cell periphery in conjuntion with integrins at the cell surface. It is known that FAK signaling can lead to changes in RNA transcription of targets involved in stem cell maintenance, DNA repair, and cell growth-survival. As biochemical cell fractionation and immunofluorescent analyses reveal that activated FAK can move to the nuclear region upon tumor cell exposure to cisplatin chemotherapy, we are testing the hypothesis that nuclear FAK has specific functions and downstreams targets promoting ovarian cancer cell survival.
PROJECT 1 – We have inactivated the FAK gene by CPRISPR DNA editing within murine and human ovarian tumor cells and have re-expressed wildtype or various function-altering mutations of FAK. The goal of these studies is to understand the role of FAK activity (it is an enzyme that will modify other proteins) as a function of its localization in cells. FAK is hypothesized to function at the cell periphery in conjuntion with integrins at the cell surface. It is known that FAK signaling can lead to changes in RNA transcription of targets involved in stem cell maintenance, DNA repair, and cell growth-survival. As biochemical cell fractionation and immunofluorescent analyses reveal that activated FAK can move to the nuclear region upon tumor cell exposure to cisplatin chemotherapy, we are testing the hypothesis that nuclear FAK has specific functions and downstreams targets promoting ovarian cancer cell survival.
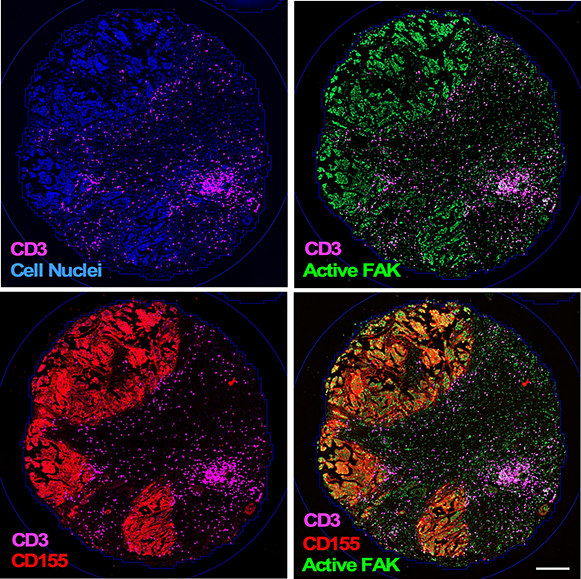
 PROJECT 2 – One of the biggest challenges to extending patient survival from recurrent ovarian cancer is to understand how these tumors can "hide" from detection by the immune system. Immunotherapy encompasses treatments that use the body's own immune system to help fight cancer. Despite success in other types of cancer, immunotherapy treatments for ovarian tumors have had limited success in promoting patient survival. Our work builds upon the idea that ovarian tumors can upregulate immune "protective" molecules and that these provide a "shield" against immune cell attack. We have found that FAK activity drives the expression of CD155 protein on tumor cells and this interacts with the TIGIT receptor on immune cells to limit immune cell activation in the tumor microenvironment.
PROJECT 2 – One of the biggest challenges to extending patient survival from recurrent ovarian cancer is to understand how these tumors can "hide" from detection by the immune system. Immunotherapy encompasses treatments that use the body's own immune system to help fight cancer. Despite success in other types of cancer, immunotherapy treatments for ovarian tumors have had limited success in promoting patient survival. Our work builds upon the idea that ovarian tumors can upregulate immune "protective" molecules and that these provide a "shield" against immune cell attack. We have found that FAK activity drives the expression of CD155 protein on tumor cells and this interacts with the TIGIT receptor on immune cells to limit immune cell activation in the tumor microenvironment.
Combination of the oral FAK inhibitor delivery with antibody-directed blockage of the TIGIT receptor significantly enhanced the survival of mice with chemotherapy resistant ovarian tumors. Mouse survival was associated with the formation of tertiary lymphoid structures within and nearby tumor sites as a marker of immune activation in the process of tumor killing. Ongoing research is focusing on the molecular mechanisms of how FAK is functioning within the tumor cells to promote CD155 expression and to determine signals and potential biomarkers involved in TIGIT and tertiary lymphoid structure regulation.
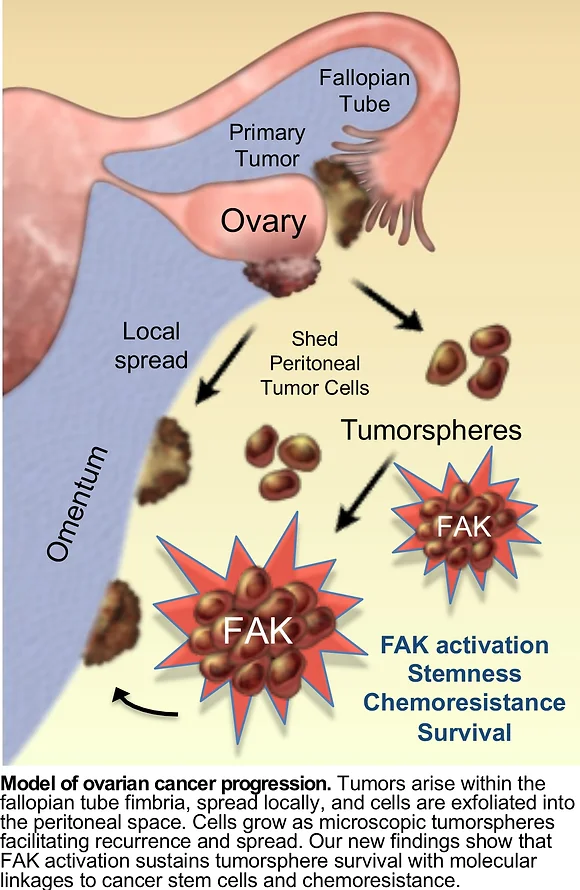 Ovarian cancer is one of the deadliest types of cancer in women. There are two main reasons for the aggressiveness of this cancer. First, ovarian cancer cells can spread to other parts of a woman’s body before she has been diagnosed, where the cells grow as tiny clumps or spheres, also called tumorspheres. Second, in the majority of patients, some ovarian cancer cells will develop resistance to the chemotherapy used. It is not clear exactly how these tumor cells become resistant to therapy. One way in which cells could do this is by gaining extra copies of genes that remove toxic substances or repair DNA, which help tumor cells withstand the chemotherapy drugs.
Ovarian cancer is one of the deadliest types of cancer in women. There are two main reasons for the aggressiveness of this cancer. First, ovarian cancer cells can spread to other parts of a woman’s body before she has been diagnosed, where the cells grow as tiny clumps or spheres, also called tumorspheres. Second, in the majority of patients, some ovarian cancer cells will develop resistance to the chemotherapy used. It is not clear exactly how these tumor cells become resistant to therapy. One way in which cells could do this is by gaining extra copies of genes that remove toxic substances or repair DNA, which help tumor cells withstand the chemotherapy drugs.
The Schlaepfer lab has developed a new mouse ovarian tumor model to study how some ovarian cancer cells resist chemotherapy. By genomic sequencing of ovarian cancer cells from mice at early and late stages of the disease, we identified a pattern of gene gains and losses that parallel gene changes occurring in human serous ovarian cancer. Aggressive cells had more genetic changes and had spontaneously acquired chemoresistance.
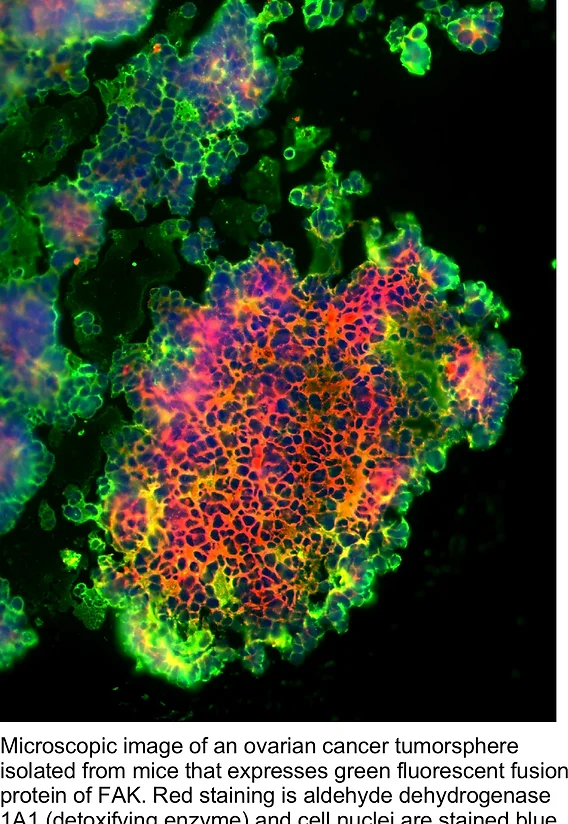 One of these changes affected the gene for a protein called FAK, which was found to have more copies than normal. The FAK protein is an enzyme (tyrosine kinase) that is best known for promoting cell movement. In cells from mice with late-stage cancer, FAK was active and present at high levels. In mouse tumors and in culture, acquired platinum resistance can facilitate ovarian tumorsphere dependence on FAK for growth.
One of these changes affected the gene for a protein called FAK, which was found to have more copies than normal. The FAK protein is an enzyme (tyrosine kinase) that is best known for promoting cell movement. In cells from mice with late-stage cancer, FAK was active and present at high levels. In mouse tumors and in culture, acquired platinum resistance can facilitate ovarian tumorsphere dependence on FAK for growth.
To understand how FAK makes ovarian cancer cells resistant to chemotherapy, we have created FAK knockout and reconstituted cells. We identified a set gene targets associated with FAK expression and a subset linked to intrinsic FAK activity in tumorspheres. Interestingly, activated b-catenin expression (a mediator of Wnt signaling and cancer stem cells) was sufficient to rescue FAK loss or inactivation phenotypes in 3D culture. However, b-catenin did not promote FAK-null tumor growth in mice. Thus, FAK selectively promotes oncogenic signaling in vivo through a yet to be defined signaling linkage.
Ongoing work will test the hypothesis that stress-induced FAK activation in tumorspheres surviving within a mouse peritoneal environment triggers specific cellular reprogramming, fostering stem-like state of heightened oncogenicity. Studies are focused on elucidating the oncogenic FAK signaling role supporting chemoresistance and we will identify other genetic changes associated with ovarian tumor aggression.
Solid tumors are comprised of a mix cells - fibroblasts, blood vessels, immune cells, as well as the accumulation of extracellular matrix proteins. Signal cross-talk between these cells within the microenvironment regulates tumor progression. Although much is known regarding how the master tumor suppressor p53 can function as a barrier to tumor progression within tumor cells, recent studies have uncovered powerful roles for cell extrinsic effects of p53 in stimulating an anti-tumorigenic microenvironment. p53 effects can be mediated through alterations in matrix protein secretion or gene expression driving elevated cytokine factors altering immune surveillance and cell senescence. Better models are needed to guide future therapeutic strategies to recapitulate key features driving a repressive tumor microenvironment.
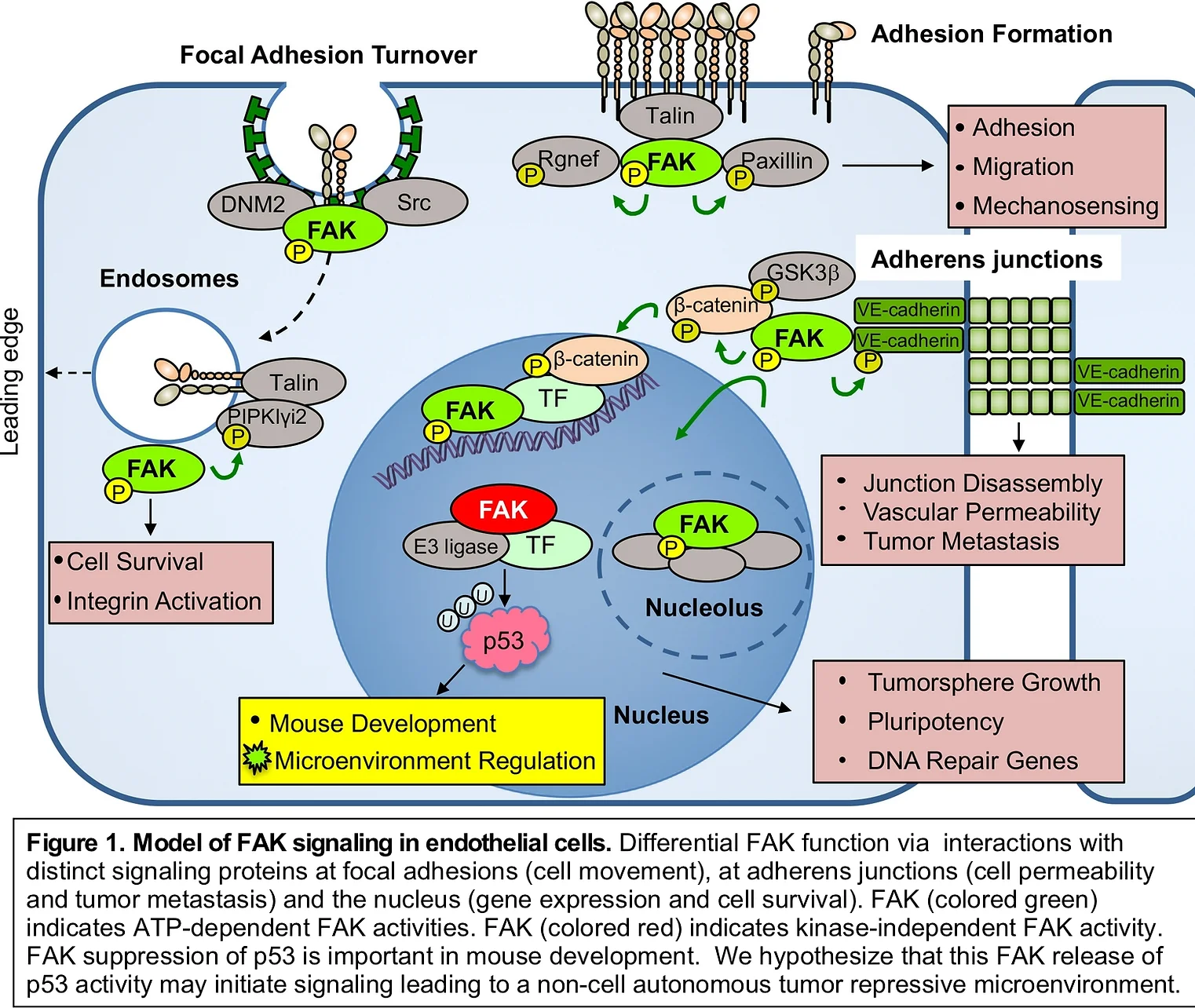
Focal adhesion Kinase (FAK) and Pyk2 are related protein-tyrosine kinases co-expressed in murine and human endothelial cells (ECs). FAK is best known for co-localization with integrins at cell adhesion sites to facilitate cell migration, mechano-sensing, and cell survival signaling (Fig. 1). In ECs, growth factors such as VEGF (vascular endothelial growth factor) can facilitate FAK activation and recruitment to cell-cell junctions in the control of vascular permeability and tumor spread. Research efforts have been focused on the role of FAK activity in signal transduction (Fig. 1, green) as small molecule FAK inhibitors are undergoing clinical trial testing. Pharmacological and genetic FAK inhibition can prevent angiogenesis in mice. However, the kinase-independent role for FAK in the nucleus as a powerful regulator of EC gene expression (Fig. 1, red) has received less research attention.
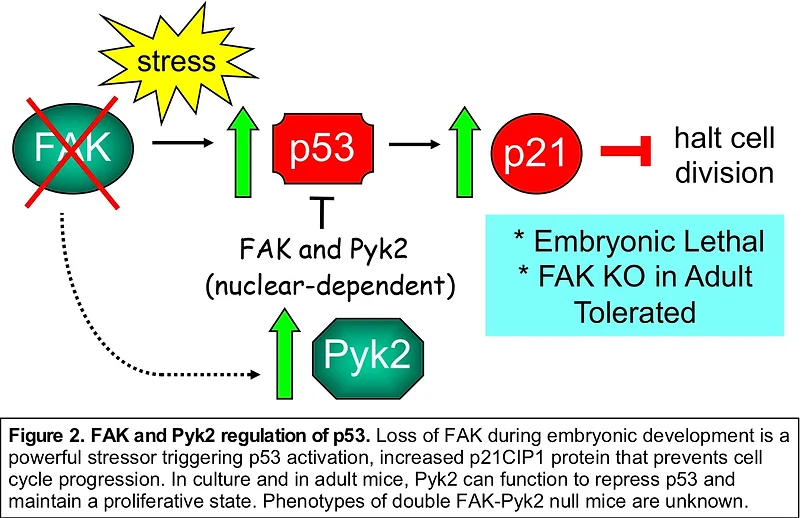 FAK knockout (KO) or kinase-inactive FAK point-mutant knockin yield embryonic lethal phenotypes in mice characterized by vascular deformation. FAK-null primary embryonic fibroblast proliferation in culture is blocked by p53 activation and released upon p53 or p21CIP1 cyclin dependent kinase inhibitor genetic inactivation (Fig. 2). EC-specific FAK (KO) is embryonic lethal and results in a genomic stress response. However, conditional FAK KO in adult mouse ECs surprisingly yields few phenotypes. We have shown that ECs possess the adaptive capacity to switch to Pyk2-dependent survival signaling upon genetic FAK inactivation. Global Pyk2 KO yields only minor phenotypes. In primary cells, FAK (or Pyk2) form a complex with p53 in the nucleus, leading to enhanced p53 ubiquitination and degradation (Fig. 1). This does not depend on intrinsic FAK activity. Targets elevated upon FAK loss remain uncharacterized.
FAK knockout (KO) or kinase-inactive FAK point-mutant knockin yield embryonic lethal phenotypes in mice characterized by vascular deformation. FAK-null primary embryonic fibroblast proliferation in culture is blocked by p53 activation and released upon p53 or p21CIP1 cyclin dependent kinase inhibitor genetic inactivation (Fig. 2). EC-specific FAK (KO) is embryonic lethal and results in a genomic stress response. However, conditional FAK KO in adult mouse ECs surprisingly yields few phenotypes. We have shown that ECs possess the adaptive capacity to switch to Pyk2-dependent survival signaling upon genetic FAK inactivation. Global Pyk2 KO yields only minor phenotypes. In primary cells, FAK (or Pyk2) form a complex with p53 in the nucleus, leading to enhanced p53 ubiquitination and degradation (Fig. 1). This does not depend on intrinsic FAK activity. Targets elevated upon FAK loss remain uncharacterized.
Ongoing work in the Schlaepfer lab has extended the importance of FAK and Pyk2 regulation of p53 equilibrium in ECs beyond development. Using a new transgenic and EC-specific conditional knockout mouse model that results in the combined loss of FAK and Pyk2 expression, we are investigating the targets and signaling linkage that results in mice with a “repressive” tumor microenvironment. The studies are hypothesized to extend beyond p53 activation in the absence of FAK and Pyk2. In addition to uncovering a new signaling pathway, this project has strong translational potential to discover key stromal regulators of tumor growth.