
Breen Church Lab
- Research
- Lab Members
- Publications
- Contact
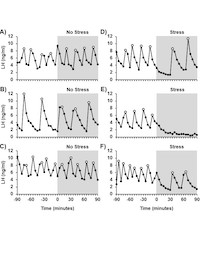 Stress is well-known to inhibit reproductive function, but the mechanisms by which various stressors alter reproductive neuroendocrine output remain incompletely defined. My research program has focused extensively on understanding the neural control of the reproductive cycle in the female, using both large and small animal models to probe the neuroanatomical and functional role of neuroendocrine sites and cellular mechanisms underlying the effect of stress on LH pulses and generation of the LH surge. Debus et al. (2002) includes our first demonstration that a stress level of corticosterone/cort (CORT) impairs pulsatile LH secretion. This study also showed that blocking CORT was not effective in reversing the effect of immune stress on GnRH/LH, which opened the door for our future work investigating mediators of a variety of stress types. In 2007, we evaluated pulsatile GnRH and LH secretion in the female sheep and determined for the first time that psychosocial stress robustly impairs the amplitude of GnRH pulses, leading to reduced LH and that an antagonist of glucocorticoid receptors could reverse the effect of stress upon the gonadotrope. The sheep is a powerful model in which GnRH and LH can be monitored simultaneously. This work has led to recent work in which my group was the first to show that psychosocial stress impairs LH pulses in female mice. Together with Dr. McCosh, we are investigating whether the pathways activated by different stress types [(i.e. metabolic (2019), immune (2020), psychosocial (2017)] are distinct in terms of signaling molecules and neural substrates.
Stress is well-known to inhibit reproductive function, but the mechanisms by which various stressors alter reproductive neuroendocrine output remain incompletely defined. My research program has focused extensively on understanding the neural control of the reproductive cycle in the female, using both large and small animal models to probe the neuroanatomical and functional role of neuroendocrine sites and cellular mechanisms underlying the effect of stress on LH pulses and generation of the LH surge. Debus et al. (2002) includes our first demonstration that a stress level of corticosterone/cort (CORT) impairs pulsatile LH secretion. This study also showed that blocking CORT was not effective in reversing the effect of immune stress on GnRH/LH, which opened the door for our future work investigating mediators of a variety of stress types. In 2007, we evaluated pulsatile GnRH and LH secretion in the female sheep and determined for the first time that psychosocial stress robustly impairs the amplitude of GnRH pulses, leading to reduced LH and that an antagonist of glucocorticoid receptors could reverse the effect of stress upon the gonadotrope. The sheep is a powerful model in which GnRH and LH can be monitored simultaneously. This work has led to recent work in which my group was the first to show that psychosocial stress impairs LH pulses in female mice. Together with Dr. McCosh, we are investigating whether the pathways activated by different stress types [(i.e. metabolic (2019), immune (2020), psychosocial (2017)] are distinct in terms of signaling molecules and neural substrates.
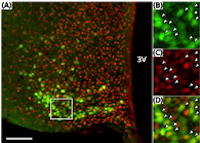 My research program has focused extensively on understanding the neural control of the reproductive cycle in the female, using both large and small animal models to probe the neuroanatomical and functional role of neuroendocrine sites and cellular mechanisms. In collaboration with Fred Karsch Ph.D., my early work investigated neuroendocrine mechanisms underlying CORT-induced reproductive suppression at both the hypothalamic and pituitary levels to impair pulsatile GnRH as well as to reduce the pituitary response to GnRH (2004). We followed this up by investigating whether the effect of CORT changes with season, as do other steroids in seasonal breeding species, such as the sheep (2006). Although the effect of CORT on the pituitary gland is not influenced by ovarian steroids (2008), we have observed the effect of CORT to impair the hypothalamus is altered by circulating levels of estradiol in the female sheep (2009). We showed that elevated CORT inhibited the GnRH pulse frequency and impaired positive-feedback to induce the GnRH/LH surge using the sheep, a powerful model in which neural secretion of hypothalamic neuropeptides can be collected and measured. This has been a research aim that we have further evaluated in the female mouse
My research program has focused extensively on understanding the neural control of the reproductive cycle in the female, using both large and small animal models to probe the neuroanatomical and functional role of neuroendocrine sites and cellular mechanisms. In collaboration with Fred Karsch Ph.D., my early work investigated neuroendocrine mechanisms underlying CORT-induced reproductive suppression at both the hypothalamic and pituitary levels to impair pulsatile GnRH as well as to reduce the pituitary response to GnRH (2004). We followed this up by investigating whether the effect of CORT changes with season, as do other steroids in seasonal breeding species, such as the sheep (2006). Although the effect of CORT on the pituitary gland is not influenced by ovarian steroids (2008), we have observed the effect of CORT to impair the hypothalamus is altered by circulating levels of estradiol in the female sheep (2009). We showed that elevated CORT inhibited the GnRH pulse frequency and impaired positive-feedback to induce the GnRH/LH surge using the sheep, a powerful model in which neural secretion of hypothalamic neuropeptides can be collected and measured. This has been a research aim that we have further evaluated in the female mouse
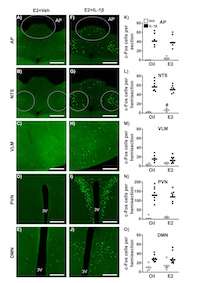 One of the goals of my research program is to understand the common and unique pathways whereby different types of stressors alter hypothalamic control of physiologic function. The female is exquisitely sensitive to alterations in ovarian steroid milieu, principally mediated by circulating levels of estradiol. This work began in the sheep in 2009, with our demonstration that the effect of CORT to impair the frequency of the GnRH/LH pulse generator was dependent on estradiol. Working with our Aussie collaborators, we showed that estradiol was also important for CORT to impair pituitary responsiveness to GnRH. Since then, we have utilized the mouse in powerful ways to probe the role of estradiol on stress-activated neurocircuitry. In 2019 we published the development of an estradiol-replacement paradigm and used this paradigm that CORT impairs LH pulsatility via suppression of KNDy cells. We just recently published our work showing that estradiol enhances the inhibitory effect of an immune cytokine (2020). Collectively, this work has begun to tease apart the physiologic regulation of estradiol-influenced stress pathways on hypothalamic neuroendocrine function.
One of the goals of my research program is to understand the common and unique pathways whereby different types of stressors alter hypothalamic control of physiologic function. The female is exquisitely sensitive to alterations in ovarian steroid milieu, principally mediated by circulating levels of estradiol. This work began in the sheep in 2009, with our demonstration that the effect of CORT to impair the frequency of the GnRH/LH pulse generator was dependent on estradiol. Working with our Aussie collaborators, we showed that estradiol was also important for CORT to impair pituitary responsiveness to GnRH. Since then, we have utilized the mouse in powerful ways to probe the role of estradiol on stress-activated neurocircuitry. In 2019 we published the development of an estradiol-replacement paradigm and used this paradigm that CORT impairs LH pulsatility via suppression of KNDy cells. We just recently published our work showing that estradiol enhances the inhibitory effect of an immune cytokine (2020). Collectively, this work has begun to tease apart the physiologic regulation of estradiol-influenced stress pathways on hypothalamic neuroendocrine function.